Molecular Mechanisms of Antibiotic Resistance in Uropathogenic Escherichia coli: A Narrative Review
Article information

Abstract
Urinary tract infections (UTIs) are among the most prevalent bacterial infections worldwide, with uropathogenic Escherichia coli (UPEC) serving as the primary causative agent. Although antibiotic therapy remains the standard of care for UTI treatment, the increasing prevalence of antimicrobial resistance has substantially reduced the effectiveness of commonly prescribed antibiotics. Resistance to trimethoprim-sulfamethoxazole (TMP-SMX), β-lactams, and fluoroquinolones is particularly concerning, as these agents constitute the principal therapeutic options for UTIs. This review examines the molecular mechanisms underlying UPEC resistance to these three classes of antibiotics, including target site modifications, efflux pump overexpression, porin regulation, and enzymatic degradation. Furthermore, it explores how these resistance determinants contribute to the development of multidrug-resistant (MDR) UPEC strains, which demonstrate cross-resistance to multiple antibiotics and present significant challenges for clinical management. Novel therapeutic strategies, such as efflux pump inhibitors, bacteriophage therapy, and genomic-guided precision medicine, are under investigation as potential solutions to address the growing global burden of MDR UPEC, alongside alternative non-antibiotic treatments. This review aims to provide a comprehensive overview of the genetic and regulatory pathways driving antibiotic resistance in UPEC, offering insights that may guide the development of effective treatment strategies and help mitigate the ongoing spread of antimicrobial resistance.
HIGHLIGHTS
Uropathogenic Escherichia coli (UPEC) is the leading cause of urinary tract infections (UTIs) worldwide, and resistance to key antibiotics such as trimethoprim-sulfamethoxazole, β-lactams, and fluoroquinolones is rapidly increasing. This resistance is driven by mechanisms including target site mutations, efflux pump overexpression, porin regulation, and enzymatic degradation. These factors contribute to the emergence of multidrug-resistant (MDR) UPEC, posing major challenges to treatment. Novel therapeutic strategies, including efflux pump inhibitors, bacteriophage therapy, and precision medicine, are being explored. A deeper understanding of resistance pathways is essential for developing effective interventions and controlling the spread of MDR UPEC.
INTRODUCTION
Urinary tract infections (UTIs) are among the most prevalent bacterial infections in humans, with approximately 150 million cases annually and a high public and financial impact [1]. Uropathogenic Escherichia coli (UPEC) causes both uncomplicated and complicated UTIs, and its virulence and adaptability allow for survival in the urinary tract despite host immune defenses [2,3]. Currently, antibiotic therapy remains a first-line therapy for UTIs, but increased resistance to most antibiotics has limited therapeutic options [4]. The most affected antibiotics include trimethoprim-sulfamethoxazole (TMP-SMX), β-lactams, and fluoroquinolones [5]. Recent epidemiologic data reveal UPEC resistance to TMP-SMX between 14.6% and 60%, and resistance to fluoroquinolones >30% in most regions [4,6]. In addition, extended-spectrum β-lactamase (ESBL)-producing and carbapenem-resistant UPEC strains have compounded the issue, with such strains being resistant to nearly all β-lactam antibiotics, including carbapenems, which are considered a last-resort option [7,8].
UPEC exhibits multiple antibiotic resistance mechanisms, including target site modifications, enzymatic inactivation, porin modifications, and regulation of efflux pump activity [9,10]. These mechanisms, often encoded on mobile genetic elements (MEGs) contribute to the rapid spread of multidrug-resistant (MDR) strains, further complicating treatment strategies [11-13]. This review aims to provide a comprehensive overview of UPEC resistance mechanisms against TMP-SMX, β-lactams, and fluoroquinolones by emphasizing genetic mutations, and shared resistance determinants. Additionally, it explores how these mechanisms contribute to MDR UPEC emergence and persistence, emphasizing the urgent need for novel antimicrobial strategies. Understanding the molecular basis of UPEC resistance is crucial for developing targeted interventions that can mitigate resistance spread and improve UTI management in clinical settings.
PATHOGENICITY AND INFECTION MECHANISMS OF UPEC
UTIs caused by UPEC occur through a complex interplay between bacterial virulence factors and host immune processes [3,14]. UPEC utilizes various strategies for colonization of the urinary tract, evasion of immune processes, and establishment of persistent infection, which results in UTI recurrence and chronicity [14,15]. These pathogenic strategies not only complicate effective treatment but also highlight a necessity for new therapeutic interventions targeting bacterial persistence and immune evasion mechanisms [3,16,17].
1. Virulence Factors of UPEC
UPEC possesses various virulence factors that facilitate adhesion to host cells, nutrient acquisition, and immune system evasion, which contribute to bacterial survival and pathogenesis [3,18]. These include adhesins, toxins, and iron acquisition systems that act synergistically to allow bacterial survival in the urinary tract [14,19,20].
A critical step in UPEC pathogenesis is the adherence to the urothelium, which is mediated by adhesion factors that enable bacterial colonization and invasion. Type 1 fimbriae (FimH), in particular, provides a crucial role in the initial attachment to bladder epithelial cells, allowing for bacterial invasion and establishment of intracellular bacterial communities (IBCs). FimH binds to mannosylated glycoproteins on bladder epithelial cells, promoting invasion and biofilm formation, contributing to recurrent UTIs. Biofilm formation creates a physical barrier that impedes antibiotic penetration, leading to insufficient drug concentrations in the deeper layers and allowing subpopulations of bacteria to remain unaffected. In contrast, adhesion in kidneys is facilitated through P fimbriae (PapG adhesin). P fimbriae recognize Gal(α1-4)Gal receptors on kidney epithelial cells, facilitating upper UTIs and pyelonephritis [21,22].
In addition to adhesion, UPEC produces toxins that disrupt host cell integrity and modulate immune responses.
Hemolysin (HlyA), a pore-forming transmembrane toxin, induces lysis of host cell, and evasion of the immune system [23-25]. Cytotoxic necrotizing factor 1 interferes with host cell signaling pathways, enhancing bacterial invasion and intracellular survival [26,27]. These virulence factors contribute to host tissue damage and bacterial survival within the urinary tract.
Furthermore, to overcome iron limitations in the urinary tract, UPEC utilizes high-affinity iron acquisition systems to compete against the host for iron. Siderophores such as enterobactin and aerobactin scavenge iron from host proteins to provides iron to support bacterial growth and virulence [28]. The expression of these iron acquisition systems is tightly regulated in response to iron availability, allowing for UPEC to efficiently adapt to the host environment [29].
2. Infection Process
The infection mechanism of UPEC is a coordinated cascade of events that enable bacterial colonization, immune evasion, and long-term persistence [16]. Following initial adhesion via type 1 fimbriae, UPEC invades urothelial cells and forms IBCs, which develop biofilm-like aggregates that protect the bacterium from immune clearance and antibiotic treatment, leading to persistent and recurrent infections [30-32]. To further evade host immune responses, the modification of lipopolysaccharides reduces the efficacy of antimicrobial peptides and complement-mediated killing of bacteria, while the expression of a polysaccharide capsule inhibits phagocytosis by immune cells [33,34]. Additionally, UPEC secretes immune-modulating factors that interfere with neutrophil recruitment and cytokine signaling, further impairing host defenses [17,35].
As the infection progresses, some UPEC cells transition into a quiescent intracellular state, forming quiescent intracellular reservoirs within bladder epithelial cells. These dormant bacterial populations evade immune responses and antibiotic treatment, but reactivate under favorable conditions to cause recurrent infections [36]. This cycle of dormancy and reactivation poses a significant therapeutic challenge, necessitating targeted strategies against persistent UPEC populations [37,38].
ANTIBIOTIC RESISTANCE MECHANISMS IN UPEC
1. TMP-SMX Resistance
UPEC employs diverse molecular mechanisms to resist commonly prescribed antibiotics. The major strategies include enzymatic inactivation, target site modification, reduced membrane permeability, and efflux pump overexpression. These mechanisms compromise the efficacy of key antibiotic classes used to treat urinary tract infection, such as TMP-SMX, β-lactams, and fluoroquinolones (Table 1).

Major antibiotic resistance mechanisms in uropathogenic Escherichia coli
TMP-SMX is a combination antibiotic that targets the bacterial folate synthesis pathway, which is essential for nucleotide biosynthesis and DNA replication (Fig. 1) [39]. Trimethoprim selectively inhibits dihydrofolate reductase (DHFR), preventing the reduction of dihydrofolate (DHF) to tetrahydrofolate (THF), a critical cofactor in DNA synthesis [40]. Sulfamethoxazole inhibits dihydropteroate synthase (DHPS), an enzyme required for the biosynthesis of DHF. By synergistically disrupting folate metabolism, TMP-SMX exerts a strong bactericidal effect against E. coli [41]. Despite its long-standing clinical use, resistance to TMP-SMX has become increasingly prevalent among UPEC strains, reducing its efficacy in UTI treatment. Epidemiological studies report substantial regional variations in TMP-SMX resistance rates, with resistance levels ranging from 14.6% to 60%, posing a major challenge to empirical treatment strategies [4]. In the United States, surveillance data from 2017 indicated that 32.1% of UPEC isolates were resistant to TMP-SMX, with resistance rates reaching up to 43.5% in certain regions [6]. Furthermore, patients with recurrent UTIs or recent exposure to TMP-SMX exhibited a significantly higher risk of resistance [42]. This increasing prevalence of TMP-SMX resistance has necessitated a reassessment of its role as a first-line treatment and stresses the urgent need for alternative therapeutic strategies.
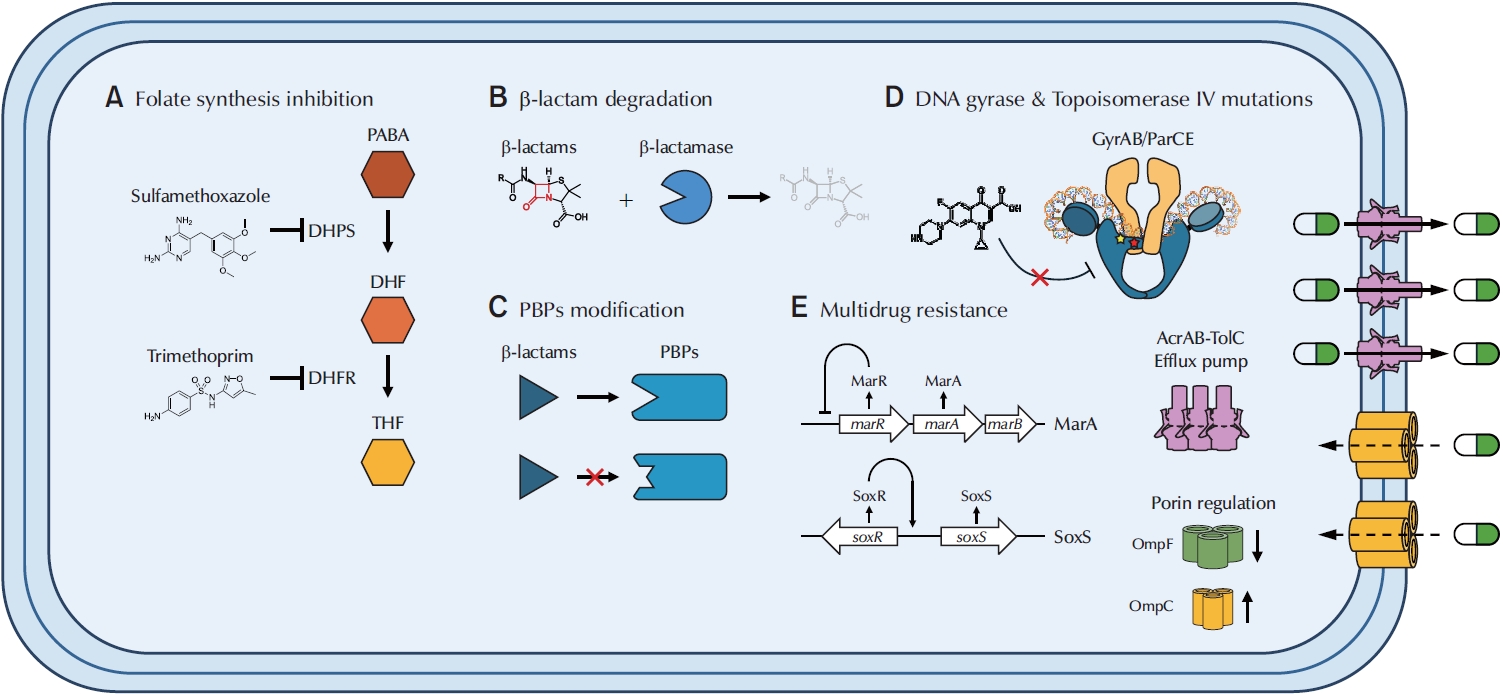
Mechanisms of antibiotics resistance (TMP-SMX, β-lactams, and fluoroquinolones) and multidrug resistance in UPEC. (A) TMP-SMX inhibits folate synthesis by blocking both DHPS and DHFR, preventing the conversion of PABA into DHF and ultimately inhibiting the reduction of DHF into THF, the active form of folate. (B) Beta-lactamases recognize and bind to the β-lactam ring of antibiotics, hydrolyzing the ring and converting it into a linear, inactive structure. (C) Point mutations in native pbp genes reduce β-lactam binding affinity, rendering antibiotics ineffective. Horizontal gene transfer of novel pbp genes can also decrease β-lactam efficacy. (D) Mutations in gyrA and gyrB alter fluoroquinolone-binding pockets in DNA gyrase, reducing drug affinity. Similarly, mutations in parC and parE modify binding sites in topoisomerase IV. (E) MarA and SoxS upregulate the AcrAB-TolC efflux system, actively pumping antibiotics out of the cell. They also downregulate expression of OmpF, a large outer membrane porin, thereby reducing antibiotic influx, while OmpC, which forms a smaller pore, is upregulated. Under normal conditions, MarR represses the marA operon; however, mutations can abolish this repression. SoxS is activated by SoxR, which may become constitutively active even in the absence of oxidative stress. TMP-SMX, trimethoprim-sulfamethoxazole; UPEC, uropathogenic Escherichia coli; DHPS, dihydropteroate synthase; DHFR, dihydrofolate reductase; PABA, para-aminobenzoic acid; DHF, dihydrofolate; THF, tetrahydrofolate; PBP, penicillin-binding protein.
The primary mechanism of TMP-SMX resistance involves the acquisition of plasmid-encoded resistance genes, encoding alternative forms of DHFR and DHPS with lowered affinities for TMP and SMX, respectively [43]. The dfrA and sul gene families play a central role in this resistance, as dfrA genes (e.g., dfrA1, dfrA5, dfrA12) encode trimethoprim-resistant DHFR variants that allow bacterial folate synthesis to continue despite TMP exposure, while sul genes (sul1, sul2, sul3) encode sulfamethoxazole-resistant DHPS enzymes, preventing SMX from inhibiting folate biosynthesis [44,45]. These genes are often carried on integrons and transposons, facilitating horizontal gene transfer and promoting their widespread dissemination among UPEC isolates [46].
In addition to plasmid-mediated resistance, chromosomal mutations in endogenous dhfr and dhps genes have also been identified in TMP-SMX-resistant UPEC isolates. These mutations alter the three-dimensional structures of DHFR and DHPS, reducing drug-binding affinity while preserving enzymatic activity [43,47]. The combination of these resistance mechanisms allows UPEC to survive despite antibiotic exposure, increasing the adaptability of this pathogen in response to selective pressure.
2. Beta-Lactam Resistance
Beta-lactam antibiotics inhibit penicillin-binding proteins (PBPs), which are important for bacterial cell wall synthesis. PBPs catalyze the cross-linking of peptidoglycan layers, a process necessary for maintaining cell wall integrity [48]. By irreversibly binding to PBPs, β-lactams disrupt peptidoglycan synthesis, leading to cell lysis and bacterial death [49]. However, UPEC has evolved multiple mechanisms to evade β-lactam activity, significantly reducing the efficiency of these antibiotics in UTI treatment.
A primary mechanism of β-lactam resistance in UPEC is the production of β-lactamases, enzymes that hydrolyze β-lactam antibiotics, rendering them ineffective. Among these, ESBLs, such as CTX-M type β-lactamase enzymes (named for their high level of activity against cefotaxime [CTX] and first observed in Münich [M]), TEM (named after a Greek patient, Temoneira, whose clinical samples yielded this gene), and SHV-type enzymes (originally reported in Klebsiella pneumoniae clinical isolate and exhibit an overall preference for hydrolyzing sulfhydryl-containing cephalosporins [hence the SHV name]), confer resistance to a broad range of β-lactams, including third-generation cephalosporins and monobactams [50-53].
MEGs play a crucial role in the rapid dissemination of β-lactam resistance in UPEC. Plasmids carrying blaCTX-M, blaTEM, and blaSHV facilitate horizontal gene transfer, promoting the cotransfer of multiple resistance determinants via integrons and transposons [54]. This accumulation of resistance genes within a single UPEC strain reduces treatment efficacy and increases the risk of therapeutic failure. Additionally, AmpC β-lactamases, often plasmid-encoded, hydrolyze cephamycins and penicillins while evading inhibition by traditional β-lactamase inhibitors such as clavulanic acid [55].
In addition to β-lactam degradation, mutations in PBPs also contribute to β-lactam resistance by reducing antibiotic binding affinity. Certain UPEC strains have mutations in both PBP2 and PBP3, leading to structural modifications that impair β-lactam binding, allowing for peptidoglycan synthesis even in the presence of antibiotics [16,56]. Another key mechanism involves the regulation of peptidoglycan remodeling, which enhances bacterial survival under β-lactam stress. Mutations in nlpD, a regulator of peptidoglycan hydrolases, have been linked to modifications in cell wall integrity, reducing the impact of β-lactam antibiotics without compromising cell viability [10,57]. This ability to alter cell wall synthesis pathways allows UPEC to tolerate β-lactam exposure while maintaining structural stability. The combination of β-lactamase production, PBP modifications, and cell wall remodeling emphasizes the complexity of β-lactam resistance in UPEC. These mechanisms allow UPEC to persist despite β-lactam treatment, contributing to recurrent infections and therapeutic challenges.
3. Fluoroquinolone Resistance
Fluoroquinolones are broad-spectrum antibiotics that target DNA gyrase (gyrA) and topoisomerase IV (parC), essential enzymes for maintaining DNA supercoiling during bacterial replication. By inhibiting these enzymes, fluoroquinolones disrupt DNA replication, ultimately leading to bacterial cell death [58]. However, UPEC has developed multiple resistance mechanisms that significantly reduce fluoroquinolone efficacy, contributing to treatment failure.
A primary mechanism of fluoroquinolone resistance is target site modification, which occurs via point mutations in the quinolone resistance-determining regions (QRDRs) of gyrA and parC. These mutations alter the structure of DNA gyrase and topoisomerase IV, reducing their binding affinity for fluoroquinolones. Among the most commonly observed mutations, S83L and D87N in gyrA and S80I and E84K in parC significantly impair fluoroquinolone binding, allowing bacterial replication to proceed despite antibiotic exposure [59,60]. Fluoroquinolone treatment can induce RecA-mediated activation of the LexA-controlled SOS response, increasing mutation rates in QRDRs and accelerating resistance development [61].
In addition to target site modifications, plasmid-mediated quinolone resistance (PMQR) genes further promote UPEC survival under fluoroquinolone exposure. Genes such as qnrA, qnrB, and qnrS encode proteins that bind to DNA gyrase and topoisomerase IV, shielding them from fluoroquinolone inhibition [62,63]. Unlike chromosomal QRDR mutations, which alter enzyme structure, PMQR genes do not modify target enzymes but instead prevent fluoroquinolones from binding effectively [64]. These genes are frequently located on conjugative plasmids, enabling horizontal gene transfer and facilitating the rapid dissemination of fluoroquinolone resistance among UPEC strains.
The presence of aminoglycoside acetyltransferase AAC(6′)-Ib-cr, a PMQR gene embedded within a gene cassette or an integron, further provides an additional resistance mechanism by modifying fluoroquinolones through acetylation. Originally identified for its role in aminoglycoside resistance, AAC(6′)-Ib-cr can also reduce the activity of ciprofloxacin and norfloxacin, further diminishing fluoroquinolone efficacy [65,66]. This dual-resistance function enhances UPEC adaptability under antibiotic pressure, contributing to multidrug resistance. The combination of QRDR mutations, plasmid-mediated protection, and enzymatic drug modification underlines the complexity of fluoroquinolone resistance in UPEC, allowing the bacterium to persist despite fluoroquinolone treatment, leading to recurrent infections and limited therapeutic options.
4. Multidrug Resistance
The emergence of MDR UPEC poses a significant challenge in UTI treatment, as these strains exhibit resistance to multiple antibiotic classes, including the aforementioned β-lactams, fluoroquinolones, and TMP-SMX. A recent experimental evolution studies on ampicillin-resistant UPEC has demonstrated that resistance to a single β-lactam antibiotic can rapidly extend to multiple drug classes, including ciprofloxacin, tetracycline, and chloramphenicol [10]. This phenomenon is primarily driven by mutations in global regulators such as marR (V66F), acrR, and envZ (T402M), which collectively lead to AcrAB-TolC efflux pump overexpression, differential regulation of porin proteins (OmpF loss and OmpC increase), and reduced membrane permeability, thus facilitating cross-resistance to unrelated antibiotics. Efflux pump overexpression and porin modifications play central roles in conferring MDR in UPEC strains [10,67,68]. Among these, the AcrAB-TolC efflux system is a key contributor to MDR phenotypes. This tripartite resistance-nodulation-division pump actively expels fluoroquinolones, β-lactams, and TMP-SMX, lowering intracellular antibiotic concentrations and reducing drug efficacy. Mutations in marR, acrR, and soxS disrupt efflux pump repression, leading to constitutive overexpression of AcrAB-TolC [10]. Additionally, overexpression of MdtK, a member of the major facilitator superfamily, has been linked to enhanced fluoroquinolone resistance in MDR UPEC isolates [69].
Alongside efflux-mediated drug extrusion, porin downregulation and structural modifications significantly contribute to MDR. The loss or mutation of outer membrane porins, particularly OmpF and OmpC, restricts antibiotic influx, thereby lowering intracellular drug concentrations [70]. These changes are often regulated by the EnvZ-OmpR 2-component system, which modulates porin expression in response to environmental stressors [10]. The combination of increased efflux activity and reduced membrane permeability creates an impermeable bacterial barrier, reinforcing MDR phenotypes and limiting the efficacy of multiple antibiotic classes [10].
CLINICAL IMPLICATIONS
MDR UPEC poses a significant challenge in the treatment of UTIs, leading to an increase in treatment failures and recurring infections. Standard first-line antibiotics, including fluoroquinolones and TMP-SMX, are losing effectiveness due to escalating resistance rates, necessitating frequent modifications to clinical guidelines [4]. This widespread resistance to first-line antibiotics limits therapeutic options, often necessitating the use of last-resort antibiotics such as carbapenems [71]. However, the rise of carbapenem-resistant UPEC strains further complicates treatment strategies and increases the burden on healthcare systems [72]. Similarly, novel β-lactamase inhibitors (e.g., avibactam and vaborbactam) are being developed to combat ESBL- and AmpC-producing UPEC strains [73,74]. In regions where TMP-SMX resistance exceeds 20%, alternative agents such as nitrofurantoin and fosfomycin are recommended [75,76].
Efflux pump overexpression plays a major role in MDR UPEC, significantly reducing the efficacy of multiple antibiotic classes. Given this challenge, clinical strategies must focus on novel approaches such as efflux pump inhibitors (EPIs) and alternative treatment methods. The use of EPIs, such as PAβN and D13-9001, is a potential strategy for enhancing therapeutic efficacy [77,78]. While EPIs have shown promise in restoring antibiotic susceptibility, their clinical application remains limited due to concerns regarding toxicity and pharmacokinetics. Further research is needed to develop safer and more effective inhibitors for use as adjunct therapies in MDR UPEC treatment.
Despite ongoing research efforts, UTI treatment remains challenging due to the formation of biofilms and quiescent intracellular reservoirs within bladder epithelial cells. These bacterial populations evade immune defenses and are protected from antibiotic exposure, making complete eradication difficult [79]. Moreover, biofilm-associated UPEC strains demonstrate heightened tolerance to multiple antibiotics, further contributing to chronic and recurrent infections [36]. Therefore, alternative treatment strategies and early diagnosis are crucial alongside antibiotic therapy. Bacteriophage therapy and immunomodulatory treatments are currently being explored as potential non-antibiotic approaches for combating MDR UPEC [80]. A study published in 2024 demonstrated the effectiveness of using lytic phages belonging to the Tequatrovirus genus to kill MDR E. coli in vivo [81]. Utilizing mouse models, the study reported that mice infected with E. coli ST131 were able to survive when treated with isolated phage cocktails. However, the current state of bacteriophage therapy and immunomodulatory treatments is still undergoing extensive testing, and it remains too early to draw definitive conclusions or determine the efficacy of other alternative therapeutic strategies for combating MDR UPEC.
The continued evolution of MDR UPEC highlights the urgent need for genomic surveillance and antimicrobial stewardship programs to track resistance trends and guide evidence-based treatment decisions [82]. Rapid molecular diagnostics, such as whole-genome sequencing and polymerase chain reaction-based resistance profiling, hold promise for more precise and personalized antibiotic selection, ultimately improving patient outcomes [83]. A comprehensive approach that incorporates novel therapeutics, advanced diagnostics, and robust infection control measures will be essential in mitigating the global public health impact of MDR UPEC.
CONCLUSIONS
UTIs remain one of the most common bacterial infections worldwide, yet their treatment is becoming increasingly difficult due to the rapid emergence of MDR UPEC [1,6,12]. As highlighted previously, resistance to first-line antibiotics such as β-lactams, fluoroquinolones, and TMP-SMX has severely limited therapeutic options, leading to higher recurrence rates and increased healthcare burdens. This review has covered the key mechanisms underlying UPEC resistance to β-lactams, fluoroquinolones, and TMP-SMX, including mutations in target enzymes, overexpression of efflux pumps, and horizontal gene transfer of resistance determinants [9-11]. Resistance to a single antibiotic, such as ampicillin, can readily extend to multiple drug classes through mutations affecting global regulators (marR, acrR, envZ), resulting in efflux pump overexpression, porin downregulation, and reduced membrane permeability [10,67,68], all of which collectively contribute to a shared mechanism of MDR to structurally unrelated antibiotics. Given these findings, addressing MDR UPEC infections requires a multifaceted approach, including the potential use of EPIs as adjunctive therapies to restore antibiotic efficacy [77,78], along with rapid molecular diagnostics and resistance surveillance. A proactive, evidence-based approach to antibiotic use and resistance monitoring will be critical in controlling the spread of MDR UPEC and improving clinical outcomes for UTI patients. Combination therapies and immunomodulatory treatments should be further explored to improve UTI management.
Notes
Funding/Support
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) [NRF-2022R1A2B5B02002256, NRF-2022R1A4A1025913, and NRF-2020M3A9H5104235 to E.-J.L.].
Conflict of Interest
The authors have nothing to disclose.
Author Contribution
Conceptualization: EJL; Data curation: NC, DUK; Writing - original draft: EJL, NC; Writing - review & editing: EJL, DUK.